

Metachromatic leukodystrophy (MLD) is a rare progressive neurological disorder that can be fatal1,2
MLD is a rare autosomal recessive lysosomal storage disorder characterized by progressive motor and neurological decline, ultimately leading to death.3,4
MLD results from pathogenic variants in the arylsulfatase A (ARSA) gene, which cause ARSA enzyme deficiency. Deficiency in the enzymatic activity of ARSA leads to an accumulation of sulfatides in the central and peripheral nervous systems, resulting in demyelination, neuroinflammation, and neurodegeneration.4,5

In the absence of newborn screening, recognizing MLD-related symptoms early is critical to obtaining an MLD diagnosis and potentially qualifying for age- and symptom-based treatment options.1,6
Presenting signs and symptoms

MLD is a spectrum disorder that is classified into clinical subtypes based on the age of symptom onset: early-onset (late-infantile, <30 months; early-juvenile, >30 months to <7 years), and late-onset (late-juvenile and adult, ≥7 years) MLD.7-10
Signs and symptoms of MLD categorized by age7-16
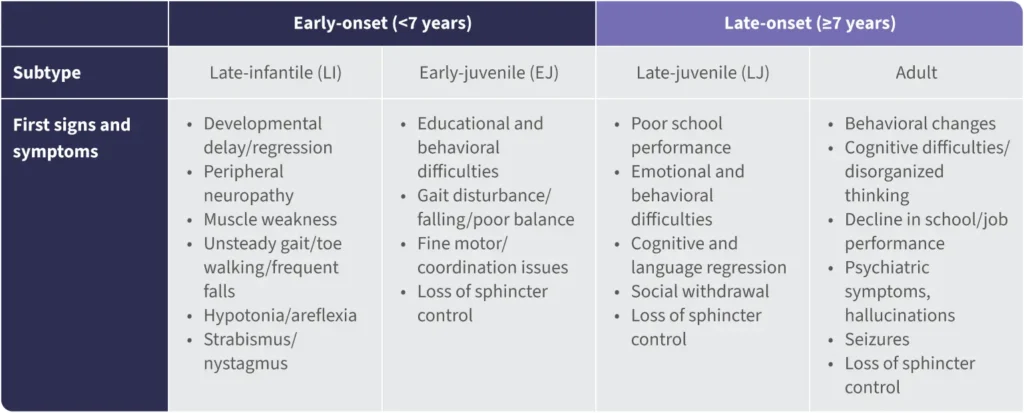
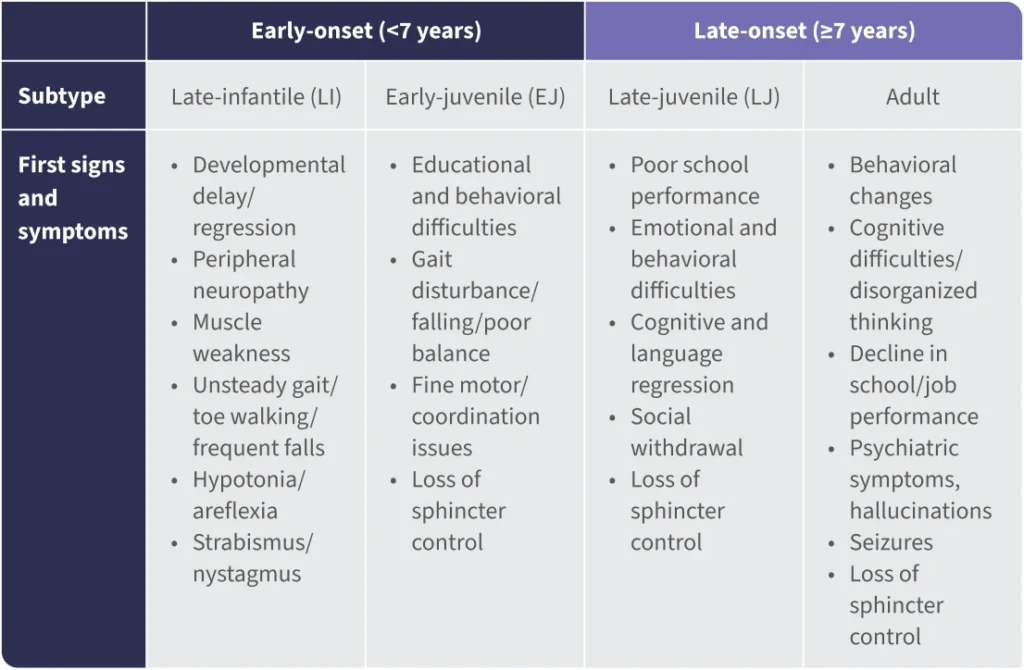
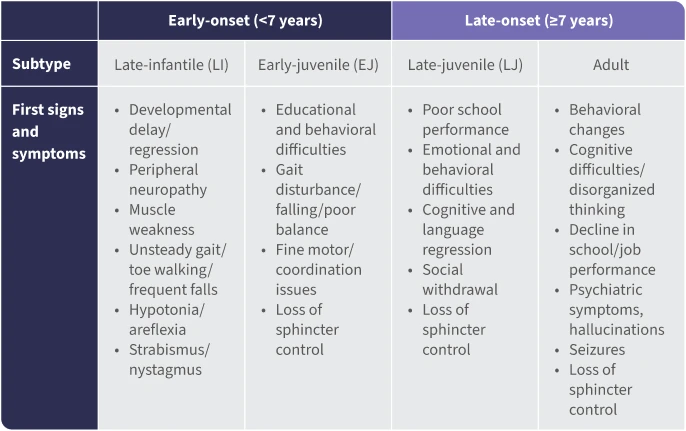

Since MLD initially presents as a constellation of nonspecific symptoms, children are often misdiagnosed.17
Early-onset MLD is the most prevalent form of MLD

>60% of all MLD cases are early-onset MLD.18

Late-infantile is the most common subtype, with rapid neurological decline starting before 2.5 years of age.1

Did you know?
Late-infantile MLD typically manifests with symptoms within the first 30 months of life and is the most aggressive and prevalent subtype, often leading to mortality within 5 years of onset.19

The first symptom of late-infantile MLD is often a plateau or delay in gross motor development or a failure to achieve independent walking.10-13,15,19

After symptom onset, rapid regression occurs with loss of previously acquired motor, language, and cognitive skills.10-13,15,19

In early-juvenile MLD, loss of independent walking typically occurs about 20 months after symptoms begin, compared with only 5 months in the late-infantile subtype.10-13,15,19

After loss of independent walking, both late-infantile and early-juvenile MLD progress with the same rapid and severe decline in motor and other functions.10-13,15,19
Early diagnosis is critical.1 Once symptoms begin, children with early-onset MLD rapidly lose all motor and cognitive skills.2,10,15
Clinical course of early-onset MLD10-13,15,19
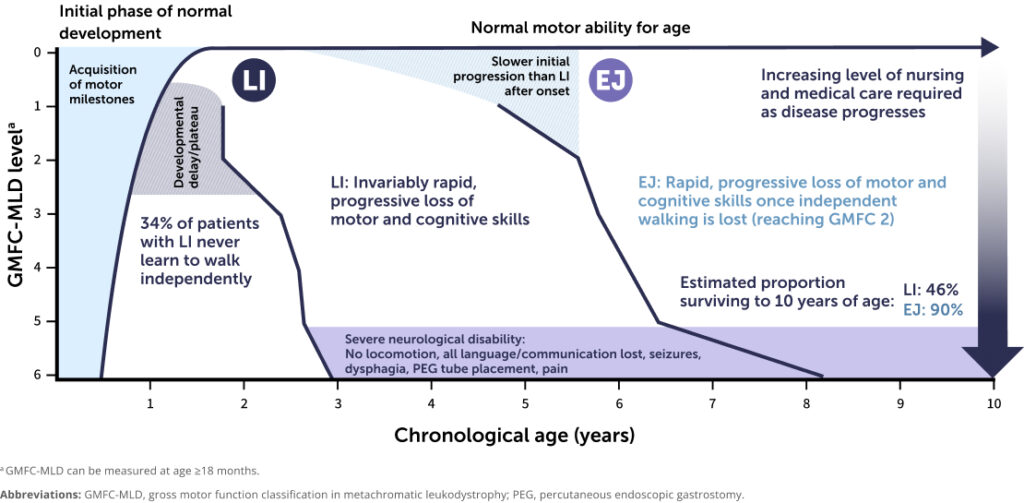
Time from symptom onset to:
Loss of ALL locomotion (years)10,15
COMPLETE loss of expressive language (years)2,10,15
Without universal newborn screening, diagnosis of early-onset MLD is challenging. If you suspect early-onset MLD, urgently refer the patient to a neurometabolic expert or leukodystrophy center of excellence for evaluation. Early identification is key to enabling treatment before the disease progresses.1
MLD is characterized by a subacute period often marked by nonspecific symptoms, such as developmental delay, gallbladder disease, and cranial neuropathies.1 Many patients with MLD, particularly those with late-juvenile MLD, present with a delay or plateau in motor milestone achievement. Clinical presentation of motor symptoms is followed by rapid neurologic regression, typically after the loss of independent ambulation.20-23
In the absence of newborn screening, it is critical to recognize MLD early.1


Access consensus guidelines for diagnosis and clinical management of MLD
Diagnostic approaches
MLD rapidly progresses, increasing the urgency for early diagnosis.11,18 Symptoms vary by patient and MLD subtype. Recognition of symptoms is essential to speed diagnosis and referrals, preventing long delays in identifying the disease.1
MLD is typically suspected based on a characteristic pattern of progressive motor and cognitive decline.15 Confirmatory diagnostic testing includes1:
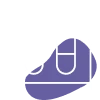
Biochemical testing
Measurement of urinary sulfatides and leukocyte ARSA enzyme activity (normal range: 21-88 nmol/mg/h) to assess metabolic abnormalities.
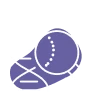
Genetic testing
Sequencing of the ARSA gene to identify biallelic pathogenic variants.
Magnetic resonance imaging (MRI) is only indicative, and MRI may appear normal in early stages of the disease.1,24
>50% of patients with MLD have gallbladder involvement, which can present as abdominal pain. Common gallbladder abnormalities include wall thickness and polyps.25
It is recommended to refer patients immediately to a specialty center, pediatric neurologist, or genetic counselor familiar with MLD, so a correct diagnosis can be obtained early enough in the course of disease for treatment eligibility.

Did you know?
Alterations in peripheral nerve myelination can be one of the earliest signs of MLD, often arising before demyelination is detectable on radiographic imaging.1
Treatments for MLD
There is currently no cure for MLD; however, certain children may qualify for treatments that can meaningfully alter the course of the disease. These include:
Stem cell transplantation1,26
- In late-onset MLD (symptom onset ≥7 years), bone marrow (stem cell) transplantation has the potential to delay or stop MLD progression, particularly when carried out before symptoms advance. A suitable donor match is needed, found through a registry or within the patient’s family
- Hematopoietic cell therapy (HCT) may be available for eligible patients with late-onset MLD who are presymptomatic or minimally symptomatic
Gene therapy1,27
- Ex vivo gene therapy is an option for early-onset MLD as clinically indicated and available. This approach does not require a donor-matched transplant since the patient’s own cells are used

Symptom management1,28
- Symptom management treatments are available to address the specific needs of children with MLD
- Comprehensive care is beneficial for all patients, including those who are already symptomatic
Resources


Learn more about clinical evidence on a treatment option for eligible patients with early-onset MLD

United Leukodystrophy Foundation
Information on MLD and resources for caregivers to access medical, social, and genetic counseling services
Calliope Joy Foundation
Supports families affected by leukodystrophies through funding research, raising awareness, and providing access to resources and medical care
References:
1. Adang LA, Bonkowsky JL, Boelens JJ, et al. Consensus guidelines for the monitoring and management of metachromatic leukodystrophy in the United States. Cytotherapy. 2024;26(7):739-748.
2. Fumagalli F, Calbi V, Natali Sora MG, et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet. 2022;399(10322):372-383.
3. Beerepoot S, Boelens JJ, Lindemans C, et al. Progressive demyelinating polyneuropathy after hematopoietic cell transplantation in metachromatic leukodystrophy: a case series. J Neurol. 2024;271(7):4028-4038.
4. Santhanakumaran V, Groeschel S, Harzer K, et al. Predicting clinical phenotypes of metachromatic leukodystrophy based on the arylsulfatase A activity and the ARSA genotype? – Chances and challenges. Mol Genet Metab. 2022;137(3):273-282.
5. Farah MH, Dali CÍ, Groeschel S, et al. Effects of sulfatide on peripheral nerves in metachromatic leukodystrophy. Ann Clin Transl Neurol. 2024;11(2):328-341.
6. Lam WKK, Ream MA, Grosse SD, et al. Evidence regarding metachromatic leukodystrophy newborn screening. Pediatrics. Published online August 12, 2025.
7. Beerepoot S, Wolf NI, Wehner K, et al. Acute-onset paralytic strabismus in toddlers is important to consider as a potential early sign of late-infantile Metachromatic Leukodystrophy. Eur J Paediatr Neurol. 2022;37:87-93.
8. Beerepoot S, Nierkens S, Boelens JJ, Lindemans C, Bugiani M, Wolf NI. Peripheral neuropathy in metachromatic leukodystrophy: current status and future perspective. Orphanet J Rare Dis. 2019;14(1):240.
9. Eichler F, Sevin C, Barth M, et al. Understanding caregiver descriptions of initial signs and symptoms to improve diagnosis of metachromatic leukodystrophy. Orphanet J Rare Dis. 2022;17(1):370.
10. Fumagalli F, Zambon AA, Rancoita PMV, et al. Metachromatic leukodystrophy: a single-center longitudinal study of 45 patients. J Inherit Metab Dis. 2021;44(5):1151-1164.
11. Wang RY, Bodamer OA, Watson MS, Wilcox WR; ACMG Work Group on Diagnostic Confirmation of Lysosomal Storage Diseases. Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med. 2011;13(5):457-484.
12. Gieselmann V, Krägeloh-Mann I. Metachromatic leukodystrophy—an update. Neuropediatrics. 2010;41(1):1-6.
13. von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrophy. In: Scriver CR, Sly WS, Childs B, et al. The Metabolic and Molecular Bases of Inherited Disease. Vol 3. 8th ed. McGraw-Hill; 2001:3695-3724.
14. Gomez-Ospina N. Arylsulfatase A deficiency. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993-2025. https://www.ncbi.nlm.nih.gov/books/NBK1130/
15. Kehrer C, Elgün S, Raabe C, et al. Association of age at onset and first symptoms with disease progression in patients with metachromatic leukodystrophy. Neurology. 2021;96(2):e255-e266.
16. Dufour J, et al. Natural history of adult-onset metachromatic leukodystrophy: findings from a large retrospective multicentric cohort. Eur J Neurol. 2023;30(S1):258-259.
17. Spinner M, Burton J, Baker J. A normal MRI leading to a delayed diagnosis of metachromatic leukodystrophy. Mol Genet Metab. 2021;132(S1):S10.
18. Chang SC, Bergamasco A, Bonnin Bisonó TA, Moride Y. A systematic review on the birth prevalence of metachromatic leukodystrophy. Orphanet J Rare Dis. 2024;19(1):80.
19. Mahmood A, Berry J, Wenger DA, et al. Metachromatic leukodystrophy: a case of triplets with the late infantile variant and a systematic review of the literature. J Child Neurol. 2010;25(5):572-580.
20. Hunter’s Hope. Metachromatic leukodystrophy. Accessed January 9, 2025. https://www.huntershope.org/family-care/leukodystrophies/metachromatic-leukodystrophy/
21. Groeschel S, Kehrer C, Engel C, et al. Metachromatic leukodystrophy: natural course of cerebral MRI changes in relation to clinical course. J Inherit Metab Dis. 2011;34(5):1095-1102.
22. Kehrer C, Blumenstock G, Gieselmann V, Krägeloh-Mann I; GERMAN LEUKONET. The natural course of gross motor deterioration in metachromatic leukodystrophy. Dev Med Child Neurol. 2011;53(9):850-855.
23. Metachromatic leukodystrophy. National Organization for Rare Disorders. Accessed June 13, 2024. https://rarediseases.org/rare-diseases/metachromatic-leukodystrophy/#symptoms
24. Schoenmakers DH, Beerepoot S, Krägeloh-Mann I, et al. Recognizing early MRI signs (or their absence) is crucial in diagnosing metachromatic leukodystrophy. Ann Clin Transl Neurol. 2022;9(12):1999-2009.
25. Adang LA, Sherbini O, Ball L, et al. Revised consensus statement on the preventive and symptomatic care of patients with leukodystrophies. Mol Genet Metab. 2017;122(1-2):18-32.
26. Schoenmakers DH, Mochel F, Adang LA, et al. Inventory of current practices regarding hematopoietic stem cell transplantation in metachromatic leukodystrophy in Europe and neighboring countries. Orphanet J Rare Dis. 2024;19(1):46.
27. Fumagalli F, Calbi V, Gallo V, et al. Long-term effects of atidarsagene autotemcel for metachromatic leukodystrophy. N Engl J Med. 2025;392(16):1609-1620.
28. Keller SR, Mallack EJ, Rubin JP, et al. Practical approaches and knowledge gaps in the care for children with leukodystrophies. J Child Neurol. 2021;36(1):65-78.